Overview
There are over 150 crystal structures of the SARS-CoV-2 main protease (Mpro) available in the Protein Data Bank, and some of them have been already used as input for molecular docking and virtual screening efforts, aiming at identifying drug inhibitors that can be used to treat COVID-19. Unfortunately, these efforts have not yet produced effective drug inhibitors for this protein. One of the main limitations of computational efforts to identify inhibitors for Mpro and other SARS-CoV-2 proteins is not to account for receptor flexibility. Receptor flexibility is a known challenge in molecular docking (Antunes et al., 2015) and there is now evidence it might be particularly important for SARS-CoV-2 proteins (Jin et al., 2020).
To address this challenge, we are running molecular dynamics simulations of SARS-CoV-2 proteins, and creating ensembles of representative conformations of these proteins in solution. In the case of Mpro, in addition to ensembles produced by simulations using two different force fields (i.e., charmm and gromos), we have also created an ensemble of experimentally-determined structures which captures most of the subtle variations observed across available crystal structures of Mpro. These ensembles of receptor conformations have been made available for ensemble docking with DINC (Devaurs et al., 2019), through the DINC-COVID webserver. In a nutshell, users can easily upload their own ligands of interest, select one of the available ensembles, and obtain the best predicted binding modes, ranked by three different scoring functions (i.e., Vina, Vinardo and AutoDock4).
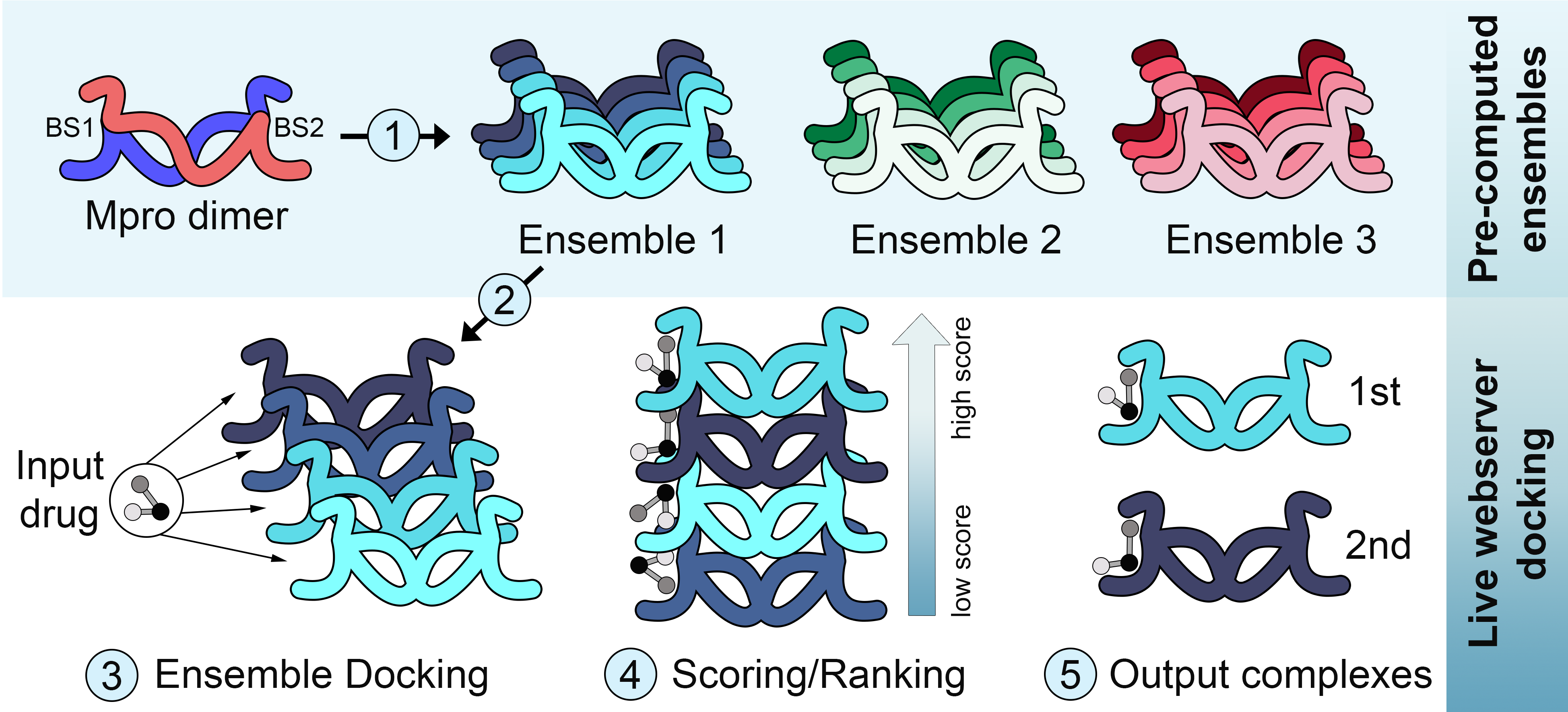
The first SARS-CoV-2 protein added to the webserver was MPro, but ensembles of other valuable targets have now been made available to users. For each SARS-CoV-2 protein of interest, a total of 10 independent 200 ns molecular dynamics simulation is executed with the GROMACS 19 package. Half of the simulations are performed with the CHARMM36 force field, and the other half with the GROMOS53a6 force field. This is an effort to avoid limitations on conformational sampling due to force field bias. CHARMM and GROMOS force fields use distinct representations for hydrogen atoms (i.e., all-atom and united-atom, respectively), which can have an impact on the results of MD simulations. Therefore, we will continue providing ensembles produced with both force fields, to address different user needs and preferences.
Conformations derived from simulations using the same force field are taken together and used to create a representative ensemble, in a process that relies on a combination of dimensionality reduction and clustering. When crystal structures are available, as in the case of MPro, they are used to create a distinct (experimentally-based) ensemble of conformations. Note that each of these ensembles also accounts for different levels of receptor flexibility. For instance, the crystal-based ensemble accounts for the least amount of flexibility, and might be better suited to identify binding modes that are similar to those observed in available crystal structures. On the other hand, the Gromos-based ensemble accounts for the largest range of receptor flexibility sampled in our simulations, and is thus best suited to identify potentially new binding modes for ligands of interest.
Algorithm
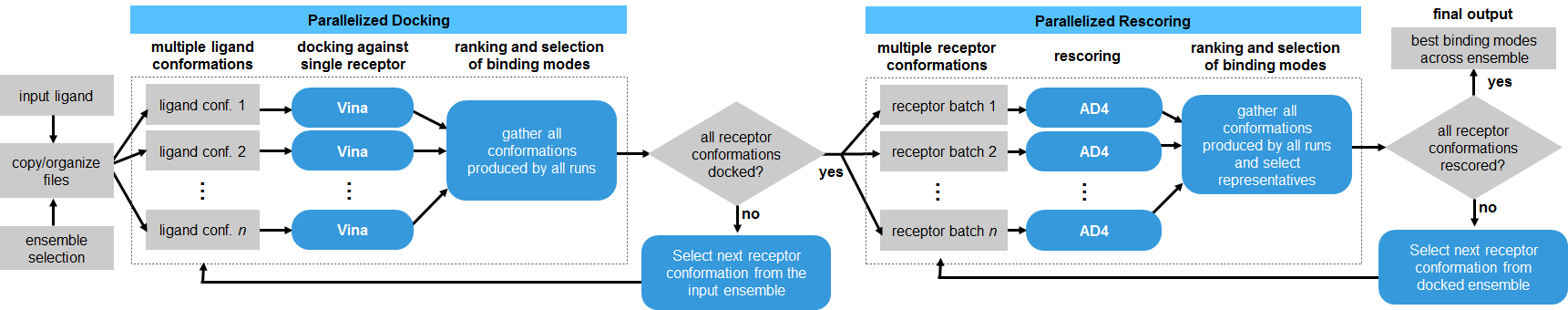
DINC is a meta-docking algorithm, in the sense that it relies on a standard docking tool, currently Vina, to perform the sampling and scoring at each docking round (Devaurs et al., 2019). Here, the DINC algorithm was adapted to performed parallelized ensemble docking for SARS-CoV-2 proteins.