Usage
Input:
- A small molecule (such as a drug or a peptide) in a .pdb or .mol2 file.
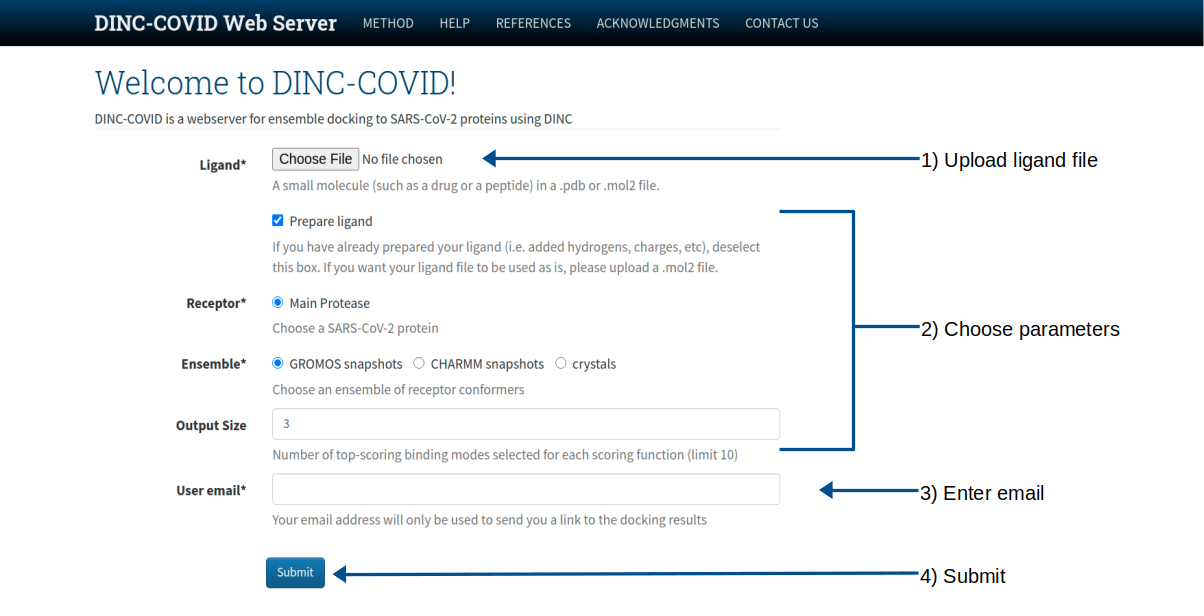
Output:
- The lowest-energy binding modes of the ligand for each scoring function (Vina, Vinardo, and AutoDock4) and the corresponding receptor conformations as PDB files
- A text file containing the energy values for the above results (dincresults.txt)
- An interactive visualization for each result using the JSMol Viewer
Sample Results
| Input file | Output |
|---|---|
| ligand | results |
| ligand | results |
Ligand Preparation
If you need so, DINC-COVID can perform the following operations on the ligand you provide.
- remove lone pairs
- add polar hydrogens and remove non-polar hydrogens
- add Gasteiger charges
Binding Sites
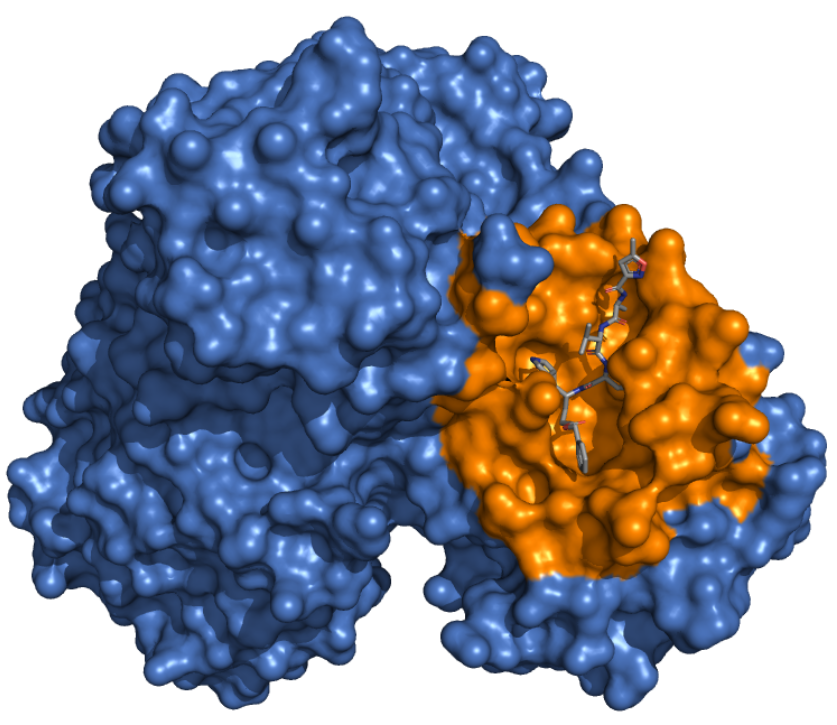 |
Mpro - Catalytic Binding Site These ensembles were built based on the dimeric state of the Mpro protein. The number of structures in the Crystal, Charmm and Gromos ensembles are 25, 14 and 11, respectively. The docking box targeting the catalytic binding site of Mpro was selected based on residues within 10 angstroms from the ligand in the crystallographic structure 6LU7. |
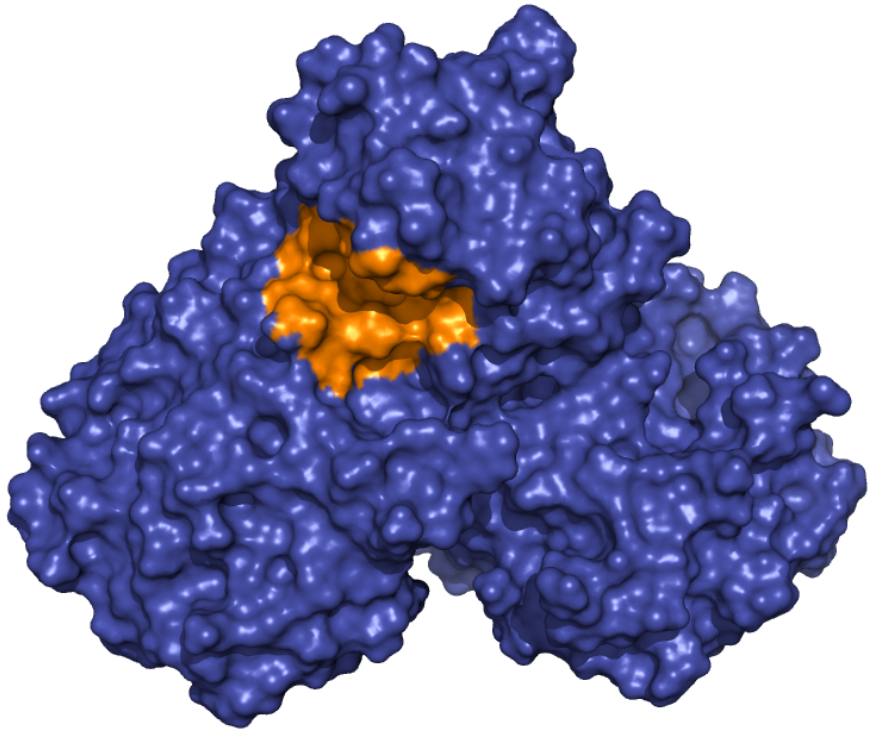 |
Mpro - Allosteric Binding Site These ensembles were built based on the dimeric state of the Mpro protein. The number of structures in the Crystal, Charmm and Gromos ensembles are 24, 10 and 9, respectively. The docking box was set based on the binding site of ebselen and comprises the area within 10 angstroms from residues Gln107, Pro108, Ile200, Val202, His246 and Phe294 in the crystallographic structure 6LU7. |
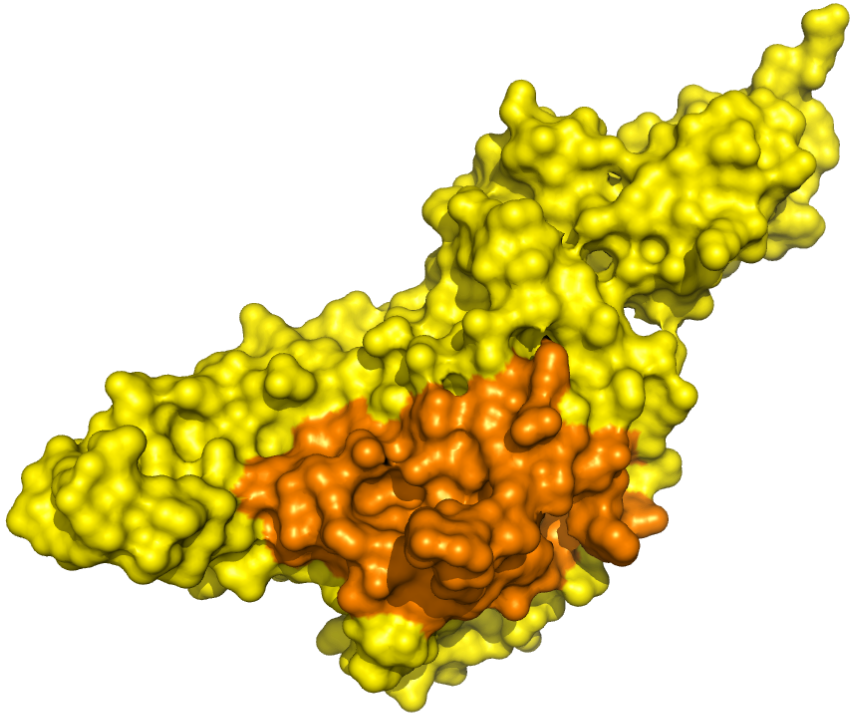 |
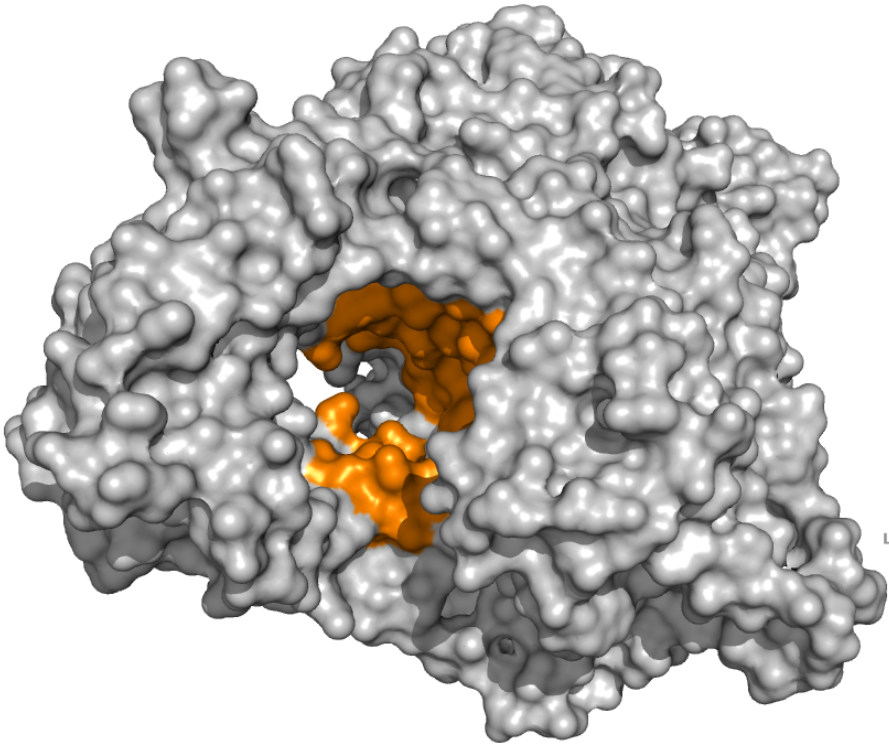
PLpro - Catalytic Binding Site These ensembles were built based on the monomeric form of the PLpro protein. The number of structures in the Crystal, Charmm and Gromos ensembles are 6, 20 and 19, respectively. The docking box targeting the catalytic binding site of PLpro was selected based on residues within 10 angstroms from the peptide inhibitor in the crystallographic structure 6WUU. |
 |
RdRp – Nucleic Acid Binding Site These ensembles were built based on the monomeric form of the RdRp protein. The number of structures in the Crystal, Charmm and Gromos ensembles are 7, 14 and 13, respectively. The docking box targeting the nucleic binding site of RdRp was set based on residues within 10 angstroms from the inhibitor remdesivir in the crystallographic structure 7BV2. |
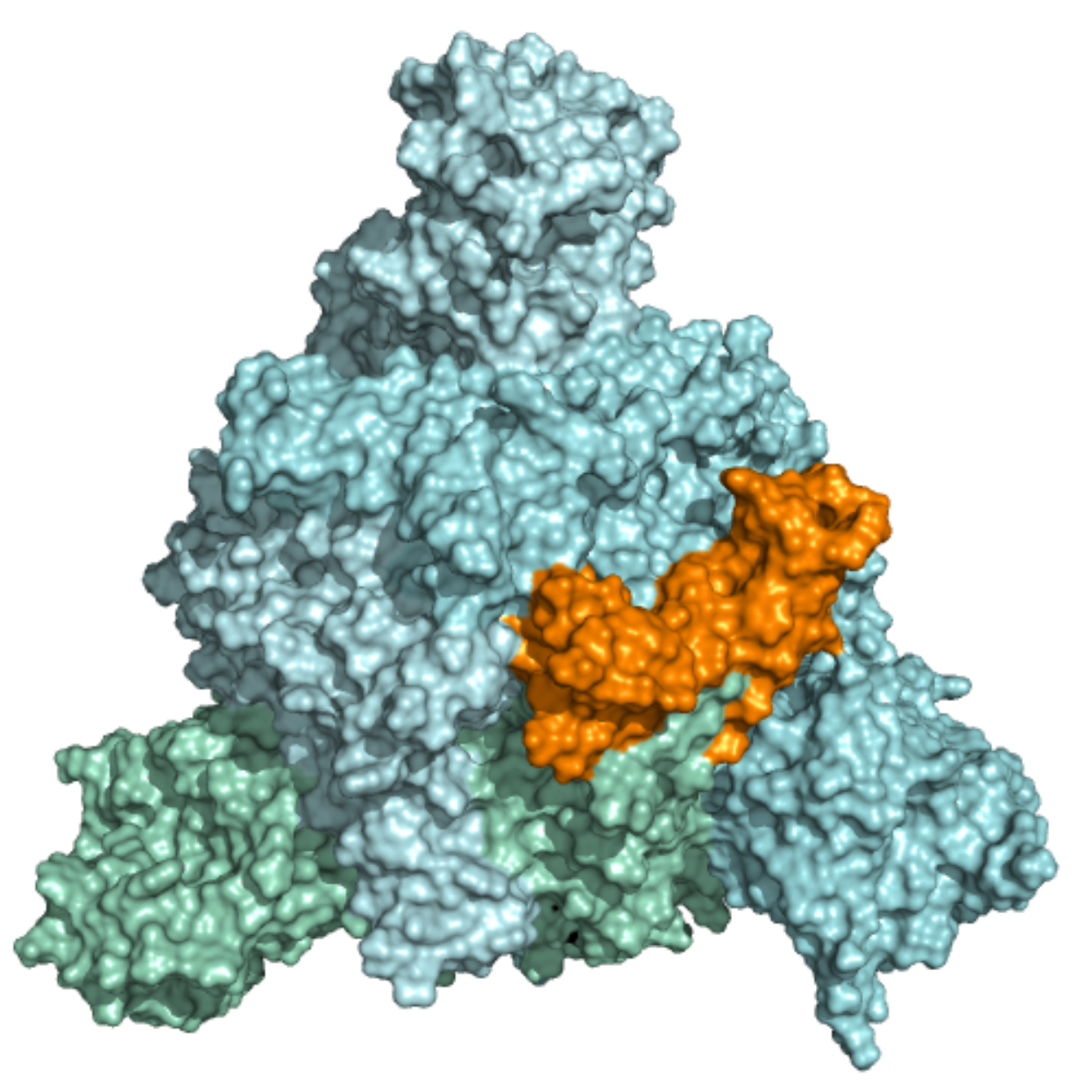 |
Spike – Receptor-Binding Motif (RBM) This ensemble was built using two 10 µs MD simulation trajectories (refs. 11021566 and 11021571)[1] of the trimeric SARS-CoV-2 spike glycoprotein with additional loop structures and glycan chains. The first trajectory was initiated in the closed state, while the second one started in a partially opened state. Further details on simulation parameters can be found at http://www.deshawresearch.com/resources_sarscov2.html. In total, 11 structures were included in this ensemble, and the docking box was selected based on the location of the RBM within the SARS-Cov-2 Spike receptor-binding domain. The RBM was observed interacting with ACE-2 (doi: 10.1038/s41586-020-2180-5) and with neutralizing antibodies (doi: 10.1038/s41586-020-2852-1). |
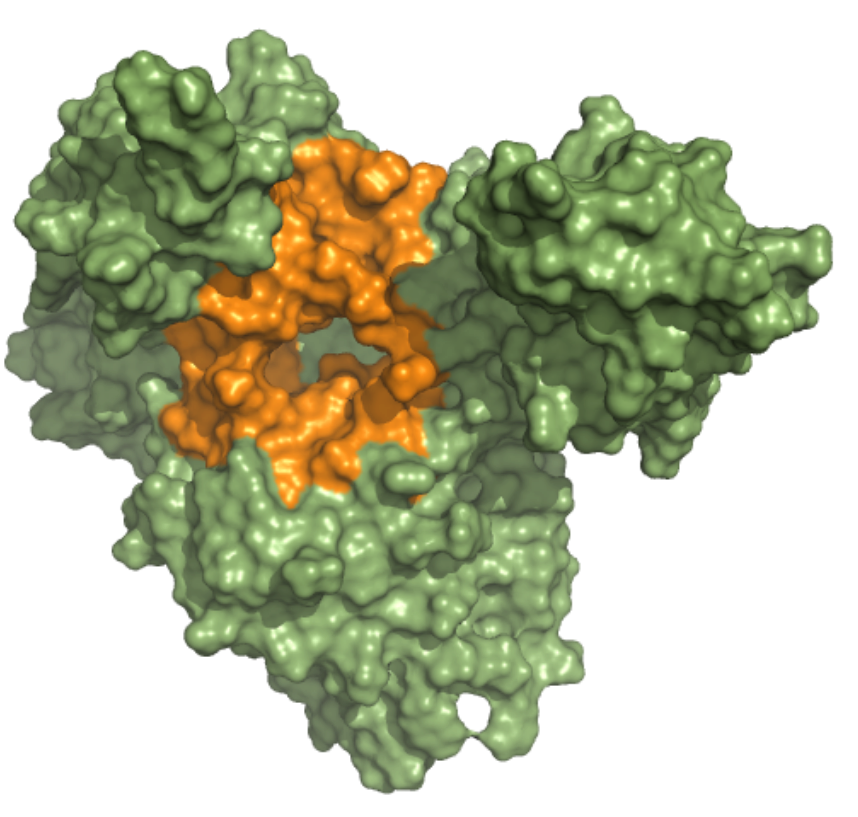 |
Helicase – Nucleic Acid Binding Site (NCB) This ensemble was built using three 5 µs MD simulation trajectories (refs. 12376233, 12376232 and 12376234)[1], conducted at 310 K in the NPT ensemble and with frames saved in intervals of 1.2 ns, of the nsp13-bound SARS-CoV-2 replication-transcription complex featuring p-RNA with a matched -1U and +1U. Further details on simulation parameters can found at http://www.deshawresearch.com/resources_sarscov2.html. This ensemble includes 18 structures, and the docking box targeting the NCB was set considering residues within 10 angstroms from nucleotide G’11 in the reference PDB structure of trajectory 12376232. |
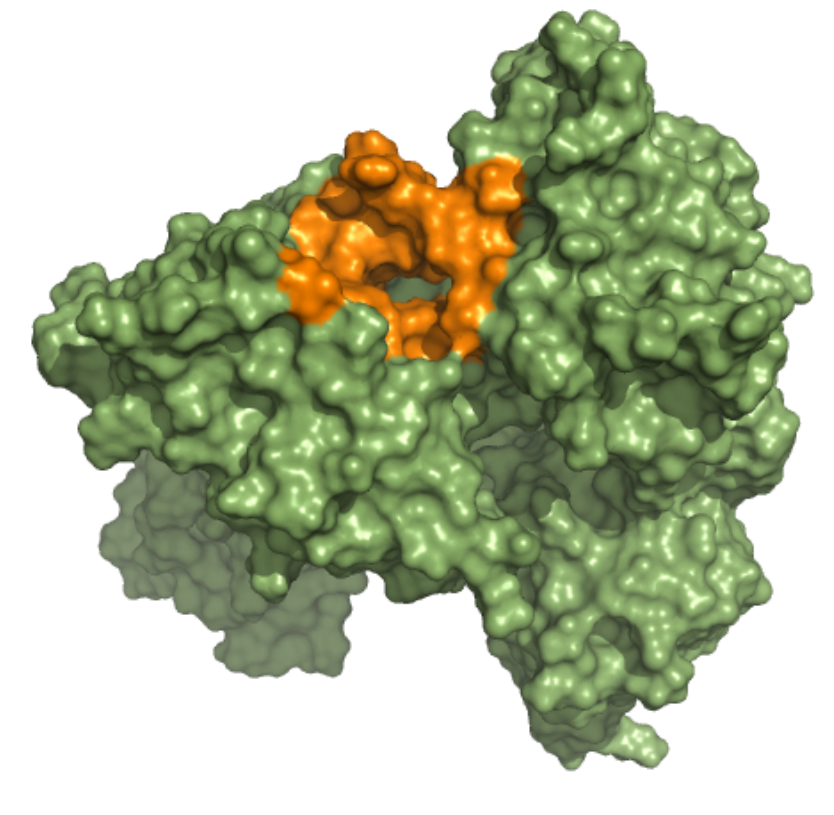 |
Helicase – ADP Binding Site This ensemble was built using three 5 µs MD simulation trajectories (refs. 12376233, 12376232 and 12376234)[1], conducted at 310 K in the NPT ensemble and with frames saved in intervals of 1.2 ns, of the nsp13-bound SARS-CoV-2 replication-transcription complex featuring p-RNA with a matched -1U and +1U. Further details on simulation parameters can be found at http://www.deshawresearch.com/resources_sarscov2.html. This ensemble contains 19 structures, and the docking box targeting the ADP binding site was set considering residues within 10 angstroms from nucleotide ADP molecule in the reference PDB structure of trajectory 12376232. |
[1] D. E. Shaw Research, "Molecular Dynamics Simulations Related to SARS-CoV-2," D. E. Shaw Research Technical Data, 2020. http://www.deshawresearch.com/resources_sarscov2.html